Protein Function Prediction
Protein function prediction involves analyzing the amino acid sequence of a protein to determine its biological roles. Accurately predicting a protein’s function provides valuable insights, allowing scientists to identify the roles of newly discovered proteins, search for proteins suited for specific tasks, or evaluate the functionality of computer-designed proteins.
Copilot provides an AI chat platform offering no-install, no-code, real-time access to advanced protein function prediction tools, enabling researchers to efficiently analyze and explore functions using a single text prompt.
ProtNLM
Predict the function of ‘DIHICGICKQQFNNLDAFVAHKQSGSQ’
| Name | Score |
|---|---|
| C2H2-type domain-containing protein | 0.54 |
| General transcription factor II-I repeat ... | 0.02 |
| Zinc finger protein | 0.019 |

ProtNLM is an ML natural language model built on T5 for predicting the name of a protein based on its amino acid sequence. More information here
Predict the function of M4332
| Name | Score |
|---|---|
| Tyrosine-protein kinase | 0.592 |
| 2.7.10.2 | 0.249 |
| Non-specific protein-tyrosine kinase | 0.019 |
AF2BIND
Predict binding sites in P32883
| resi | p(bind) |
|---|---|
| 13 | 0.8182 |
| 15 | 0.8126 |
| 16 | 0.811 |
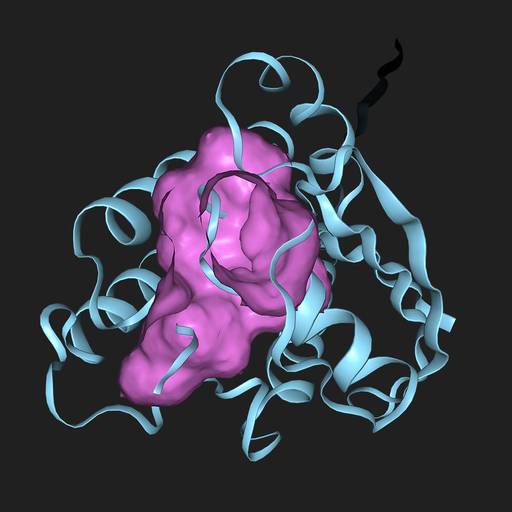
AF2BIND predicts which amino acids are part of binding sites in the protein. More information here