Protein Structure Prediction
Protein structure prediction is the process by which linear chains of amino acids and predicting its three-dimensional structures. The “folding problem” has historically posed significant challenges in molecular biology, as accurately predicting these stable forms has been complex and time-consuming, often requiring extensive experimental methods like cryo-EM or crystallography.
Copilot leverages AI to deliver tools that analyze amino acid sequences and predict their 3D structures. Requiring no installation or coding, researchers can initiate predictions seamlessly with a single text prompt.
AlphaFold
Fold ‘DIHICGICKQQFNNLDAFVAHKQSGSQ’
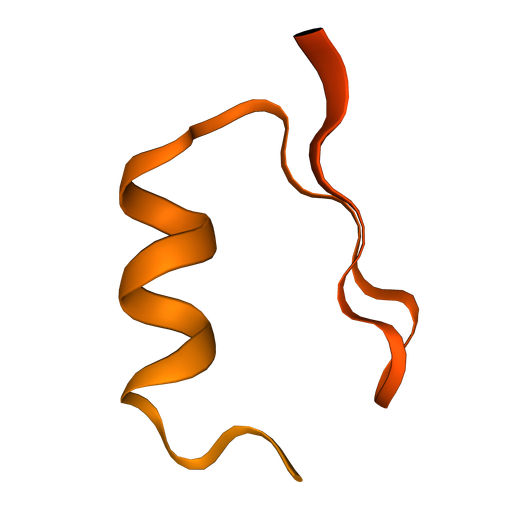
AlphaFold (aka AlphaFold2, AF2) is a transformer-based AI model for predicting the 3D structure of proteins from sequence. More information here.
ESMFold
Fold A0A7S7MT40
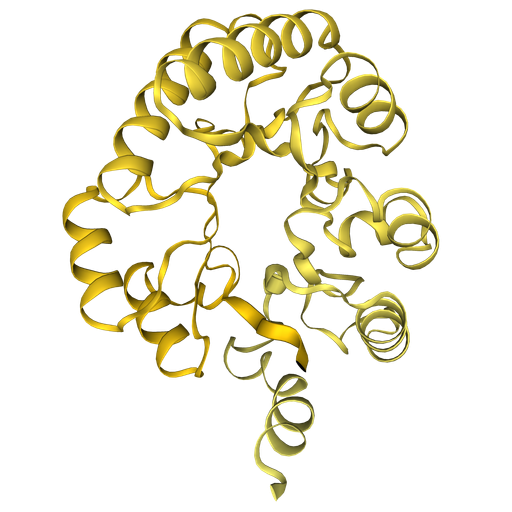
ESMFold is an ML model that is a variation of AlphaFold2 that offers higher speed. More information here.