Molecular Docking
Molecular docking predicts how molecular structures, such as proteins and ligands, interact to form stable complexes. This process has been challenging in drug design and molecular biology, often requiring extensive computational methods.
Copilot simplifies this with an AI chat platform that provides no-install, no-code, real-time access to advanced molecular docking capabilities. With a single text prompt, researchers can quickly model and analyze molecular interactions.
DiffDock
Dock asparagine into A0A1L9RXX7
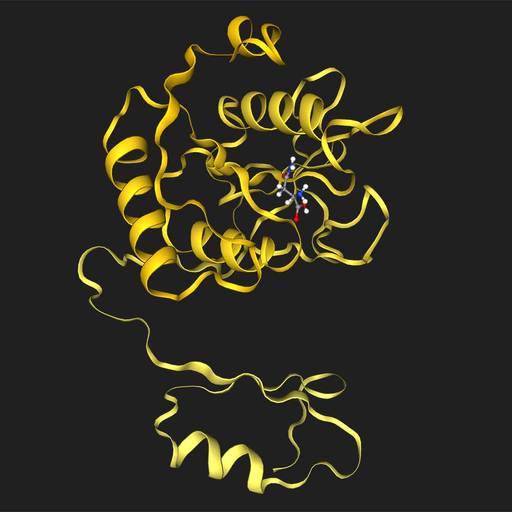
DiffDock is a diffusion-based ML model for the docking of a protein to a small molecule. More information here
AlphaFold3
Dock biotin into P22629
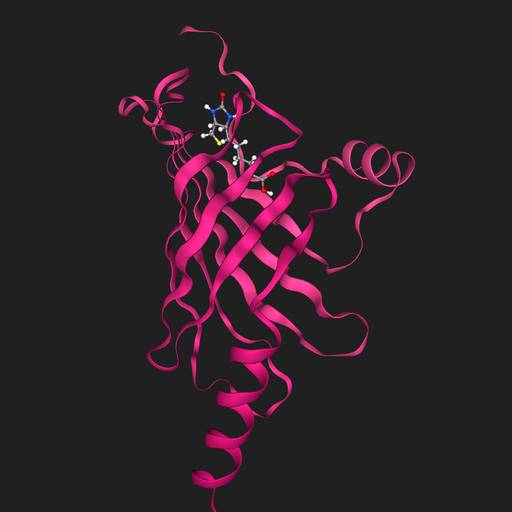
AlphaFold3 (aka AF3) is for predicting the 3D structure of proteins + nucleic acids + small molecules + post-translational modification. It is capable of multi-chains and therefore is also a cofolding or docking method. More information here