ESMFold for Protein Folding
ESMFold is a machine learning model that predicts protein 3D structures from amino acid sequences using transformer-based neural networks. Developed by META (formerly Facebook Research), it leverages evolutionary data for fast and accurate predictions.
How to Use ESMFold
Fold ‘DIHICGICKQQFNNLDAFVAHKQSGSQ’
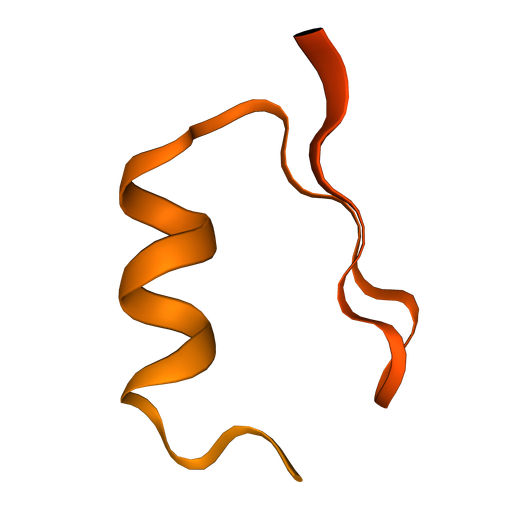
Inputs
- Protein Sequence
A string representing the single-letter amino acid sequence, consisting of the common 20 amino acids. This input is intended for single-chain (monomeric) proteins only.
Outputs
- PDB file
File containing the predicted 3D coordinates of each residue in the input sequence.
- pLDDT Scores
Specified in the b-factor column of the PDB file. A per residue confidence score between 0 and 100 with higher being better.
Examples
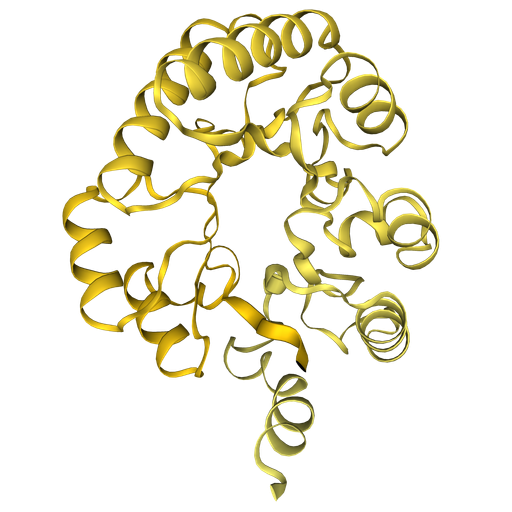
Fold A0A7S7MT40
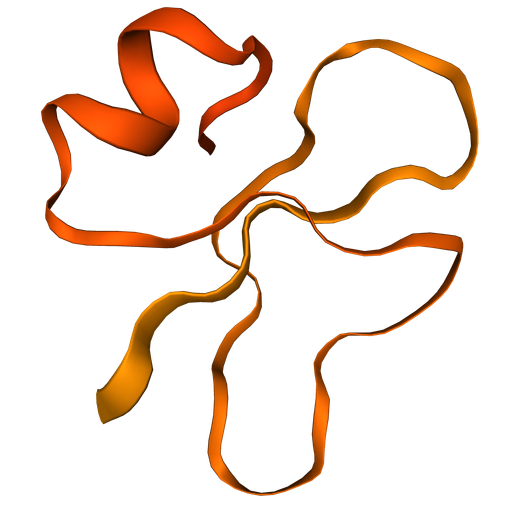
Fold 'GIGDPVTCLKSGAICHPVFCPRRYKQIGTCGLPGTKCCKKP'
Evaluating ESMFold Predictions
pLDDT Scores: The predicted local distance difference test (pLDDT) is a per-residue confidence score. Scores range from 0-50 (very low, indicating disorder/flexibility) to 90-100 (very high, suggesting highly structured/stable conformations). Understanding these scores is crucial for assessing the reliability of the predicted structure.
PAE Scores: Predicted aligned error (PAE) measures confidence between residue pairs. Scores of 0-5Å indicate high confidence (known relative positions), while scores of 20Å+ indicate low confidence (unknown relative positions). Analyzing PAE scores helps evaluate the structural alignment and potential flexibility of protein regions.